
肌萎缩侧索硬化的临床分型
北京协和医院 作者:李晓光 崔丽英
肌萎缩侧索硬化(Amyotrophic lateral sclerosis,ALS)是一种进展性神经系统变性疾病。由于上、下运动神经元丢失导致球部、四肢、胸部肌肉逐渐无力和萎缩,动眼肌及括约肌不受累。发病率约1.5/10万,患病率4-6/10万。隐袭起病,进展缓慢,呼吸衰竭死亡。发病年龄平均55岁,发病起计算平均存活3.5年。50%患者平均存活时间为2.5年。5年后20%患者存活,10年后10%存活。球部起病者存活期约2.2年,存活很少超过5年。一般发病年龄越早,存活时间越长。目前尚无治愈的方法,但近年来研究表明有许多措施可以延长患者存活期,提高患者的生活质量。为了科学合理地治疗ALS,本文就其临床分型,分期,治疗模式及对ALS病情进展的评估和随访方法进行了介绍。
一、ALS的定义及分类
ALS在一些国家有不同的名称,在法国也称为Charcot病,为纪念1869年首次描述这一疾病的马丁夏科医生;在英国称为运动神经元病,强调该病归属的类别;在美国公众称为Lou Gehrig病,这是以患此病并使此病引起公众注意的著名棒球手的名字命名的;专业杂志遵循国际神经病学联盟的命名称为ALS。我国一般将ALS和运动神经元病混用(近年来国内非医学媒体还称其为渐冻人症,源于台湾运动神经元病协会进行科普宣教所使用的俗称)。建议国内同行统一采用ALS为该病的病名,便于和国际交流。
二、ALS的诊断
1990年,由世界神经病学联合会在埃斯科里亚尔(El Escorial,西班牙)举办了一次研讨会,制定了第一个共识文件[3]。其核心为将病变累及的神经系统区域分为颈区、胸区、腰骶区、延髓区,根据所制定标准,在临床怀疑ALS的患者确诊ALS需要同时存在下运动神经元变性临床表现及上运动神经元变性的临床表现和逐步向身体其他区域扩展,且没有可以解释其临床表现的其他疾病的依据。电生理检查可以用来确认某一区域的下运动神经元损害,如每个身体区域至少两个不同的脊神经根和周围神经支配的肌肉同时存在纤颤电位和长时程的巨大运动单位电位和运动单位募集减少。根据神经功能缺损的广泛性,制定了4个诊断信度级别:肯定的ALS(在3个区域同时存在上运动神经元和下运动神经元损害表现),很可能的ALS(两个区域存在上运动神经元和下运动神经元损害表现,有些上运动神经元体征位于下运动神经元体征的头端),可能的ALS(一个区域存在上运动神经元和下运动神经元损害的体征或至少有两区域存在上运动神经元损害的体征),可疑的ALS(至少有两个区域存在下运动神经元损害的体征)。
1998年为提高灵敏度,修订了这些标准。在修订的标准(或Airlie House标准)[4]中,认为在腰骶和颈椎区域不同的脊神经根和周围神经支配的至少两块肌肉和在胸椎和头颅区域至少一块肌肉存在急性失神经支配和慢性失神经支配的表现,电生理检查可确定下运动神经元损害。修订的El Escorial标准保留3个诊断信度级别:临床肯定的ALS,很可能的ALS,可能的ALS。在电生理学检查时发现患者两个区域存在下运动神经元损害的表现,在一个区域存在上运动神经元和下运动神经元体征或一个仅有区域上运动神经元体征时,添加了临床实验室支持的很可能的ALS这个级别。去除了“可疑的ALS”级别。
因为灵敏度低及从发病到符合参加临床试验的资格需要很长一段时间,两套诊断标准一直受到批评。2006年,在淡路市召开的共识会议(日本)进行了两个修订:在确认失神经时,下运动神经元损害的电生理证据(活动性及慢性去神经征)等同于临床的下运动神经元体征,和束颤电位等同于纤颤电位和正锐波。从本质上讲,针电极肌电图被认为是一种临床检查的延续,并维持了前一版诊断标准的一般原则。其结果是临床实验室支持的很可能ALS这一级别变得多余,进一步简化了诊断标准的应用。要特别重视针肌电图对慢性神经性变化和肌束震颤的评估,留意没有纤颤或正锐波的肌肉,可改善ALS早期诊断的灵敏度。这对早期诊断,对提高进入临床试验的时机和人数,减少延迟诊断,避免延误治疗等等均有重要意义[5]。
因此,目前ALS的诊断信度级别分三级:临床肯定的ALS,临床很可能的ALS,临床可能的ALS。
三、ALS的临床分型
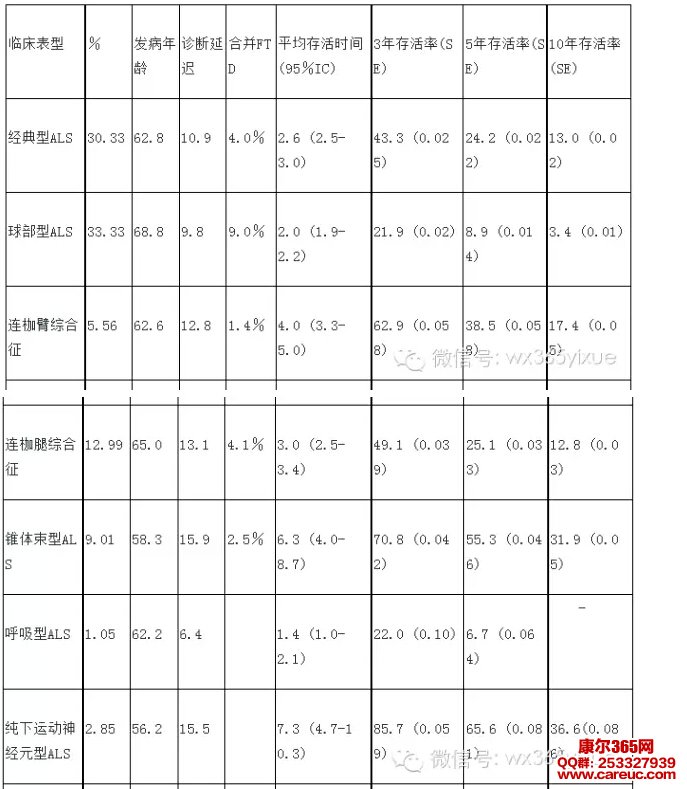
在确定ALS诊断后,要及时告知患者及家属该病的预后,这就需要对ALS自然病程、对病情发展评价方法的充分了解;在随诊时通过对患者病情变化的观察,进一步确定预后和分型。ALS通常分ALS,进行性延髓(球)麻痹,脊髓性肌萎缩和原发性侧索硬化等四类临床亚型。但研究证明有些患者的表现符合ALS的诊断标准,但以这四个亚型分类难以准确概括患者的病情发展与损害分布特点,采用新的分型有助于进一步判断患者预后及在进行临床药物试验时选择合适患者入组。近年研究主要将ALS分八个临床表型,这些类型在发病年龄,延迟诊断的时间,合并额颞叶痴呆的比率,生存期,3、5、10年存活率等均有差异(表1)[6]。
这8个分型建立诊断时的临床表现基础上,但在随访中要收集患者所有可用的资料,不断修订。⑴、经典(夏科)型ALS(Classic(Charcot)ALS ,C-ALS):在上肢或下肢出现特征性症状或体征,锥体束征明确,但并不突出。⑵、延髓型ALS(Bublar ALS,B-ALS):这些患者为延髓发病,有构音障碍和/或吞咽困难,舌萎缩,肌束震颤。在发病后的前6个月内没有脊髓损害症状。在前6个月锥体束征可以不明显,但之后要显而易见。⑶、连枷臂综合征(Flail Arm Syndrome,FA-ALS):本类型患者的特点是逐渐发展,主要是上肢近端无力和萎缩。此类型包括病程中某一阶段患者上肢的病理性深部腱反射或霍夫曼征,但无肌张力增高或阵挛。在发生症状后局限于上肢的受累功能至少有12个月。⑷、连枷腿综合征(Flail leg syndrome,FL-ALS):患者特点是逐渐进展,下肢远端出现的无力和萎缩。此类型包括病程中某一阶段患者下肢的病理性深部腱反射或巴彬斯基征,但无肌张力增高或阵挛。患者下肢近端起病的萎缩和无力,在无远端受累时列为经典型ALS。⑸、锥体束征型ALS(Pyramidal ALS,P-ALS)或(上运动神经元损害突出的ALS,upper motor neuron dominate ALS):这些患者的临床表现主要是锥体束征,主要表现为严重的痉挛性截瘫/四肢瘫,有一个或多个体征:巴宾斯基或霍夫曼征,腱反射亢进,下颌阵挛性抽动,有构音障碍和假性球麻痹。痉挛性麻痹可以存在于发病初期或疾病晚期。这些患者在发病时可以至少在两个不同区域同时表现有明显的下运动神经元损害的体征如肌肉无力和萎缩,肌电图检查存在慢性和活动性的失神经损害。⑹、呼吸型ALS(Respiratory ALS,R-ALS):这些患者发病时表现为弥漫性呼吸功能损害,为休息或在劳累时端坐呼吸或呼吸困难,在发病第6个月后只有轻微的脊髓或延髓体征。这些患者可以表现出上运动神经元受累的表现。⑺、纯下运动神经元综合征(pure lower motor neuron phenotype,PLMND或Progressive muscular atrophy, PMA):这些患者有逐渐进展的LMN受累的临床和电生理证据。这个类型中排除了①以标准化神经节段传导研究存在运动传导阻滞者,②临床上有UMN体征者,③类运动神经元病综合征疾病史者,④有家族病史的脊髓性肌萎缩症,⑤SMN1基因的缺失者或⑥CAG患者雄激素受体基因重复异常扩展的遗传性延髓脊髓性肌萎缩症,⑦神经影像学研究除外结构损害。⑻、纯上运动神经元综合征(pure upper motor neuron phenotype, PUMN或Primary lateral sclerosis PLS):这些患者上运动神经元损害的临床症状包括严重的痉挛性截瘫/四肢瘫,巴宾斯基征或霍夫曼征,反射极度活跃,下颌阵挛性抽动,构音障碍和假性球麻痹。这个类型中排除了①随访过程中按照埃斯科里亚尔标准有临床或肌电图表现的下运动神经元受累征象的患者,②类运动神经元病综合征病史患者,和③有痉挛性截瘫/四肢瘫家族史的患者和④基因突变相关的遗传性痉挛性截瘫(SPG3A,SPG4,SPG6,SPG7和SPG20)的患者。
表一ALS临床表型与存活预测(ALS各临床表型与发病年龄,诊断延迟时间,合并FTD的比例,平均存活时间,3,5和10年存活率)

四、ALS的临床分期
目前ALS已有各种临床表型和诊断信度标准,应用较广。但疾病分期标准尚不成熟,为了相对客观准确判断ALS的预后,对疾病发展情况进行划分有利于判断药物临床试验的效果及对不同阶段的患者进行有益的干预。
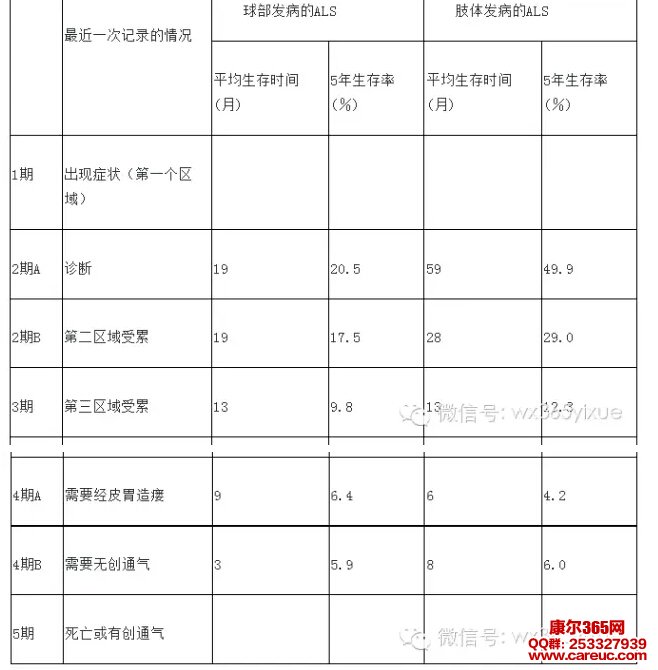
英国Roche等根据病程中最近一次患者受累的区域不同,将ALS的病程从发病到死亡分为5期。第1期为出现症状(第一个区域)。第2期A为诊断确定时,第2期B为第二区域受累。第3期为第三区域受累。第4期A为患者需要经皮胃造瘘,第4期B为患者需要无创通气(无创通气)。第5期为死亡或进行机械通气。各期平均存活时间及5年生存率均不同[7](表二)。
而意大利Chio等提出米兰都灵ALS分期系统,这个分期将患者功能分为4个域,通过每个域功能损害评分的总和来确定分期,0期为无功能丧失。第1期为一个功能域丧失。第2期为两个功能域丧失。第3期为三个功能域丧失。第4期四个功能域丧失。第5期为死亡或进行机械通气。该研究为前瞻性研究,按病程分期观察了118例ALS患者12个月的进展情况。发现分期与生活质量及神经功能,医疗费用密切相关[8](表三)。根据该系统还可预测患者生存时间[9]。
这两个分期系统各有优点。米兰都灵分期基于ALSFRS,后者是大多数ALS专家熟悉的评分量表,广泛应用于临床药物试验和日常工作,其结合了与患者功能相关的临床节点(如诊断,第二,三区域损害)或两个特定功能的丧失(需要胃造瘘或无创呼吸机)。而伦敦分期基于临床病程节点如起病症状,诊断及后续进展等,验证了地区ALS专科中心的1471例患者。
表二、伦敦ALS分期与生存期
表三,肌萎缩性侧索硬化米兰-都灵分期(ALS-MITOS)

五、ALS的临床评估
ALS目前尚无治愈办法。准确、客观、科学的评估患者病情及神经功能状态对了解疾病进展程度和对临床药物治疗试验的疗效就显得极为重要。
5.1、存活[14]。将ALS患者的存活作为临床药物试验观察指标无疑是合理的,但随着侵入或非侵入通气支持技术与营养支持的发展和应用使得这一指标受到质疑。因此,如果将存活作为终点观察指标,设计新药的临床观察方案必须考虑NIPPV和PEG的因素。
5.2、运动系统特定损害的测定。临床常用的手工运动测定(MRC评分)虽然应用广泛,但有主观因素影响且无法标准化,因此推荐用定量肌力仪肢体肌力测定。球部功能尚无理想的评估办法。前角细胞功能可用电生理对运动单位数目进行评估。上运动神经元功能常根据电生理测定(如经颅磁刺激)和功能性影象学测定。
5.3、综合测定。通常应用综合量表进行评估,目前常用的有功能评估量表(ALS-FRS)及生活质量量表(ALSAQ-40)等。
ALS功能评分量表(ALS-FRS)主要由四个球部-呼吸功能、两个上肢功能(用餐具和穿衣)、两个下肢功能(走路和爬楼梯)及两个其他功能(穿衣及洗漱、床上翻身)组成。其简便、容易操作、应用广泛,其敏感性、可靠性和稳定性已得到广泛确认。近年量表进行了改良,增加了呼吸功能的评分强度,还适用于呼吸支持的使用者。目前改良的ALS功能评分量表总分48分[16]。
目前基于书面的ALSFRS版本应用广泛,且已开发了基于网络,电话的版本,均得到验证,患者自评的得分与医生评分高度一致,这是因为书面ALSFRS版本医生的评分均基于患者对自身神经功能的描述[17]。Prize4组织基于ALSFRS斜度变化预测模型的外包竞赛,结果十三组参赛者中两组胜出,优于组织者开发的预测模型,与参加临床试验的患者病情的实际进展高度一致。组织者认为这个模型可以降低20%的临床药物试验入组患者人数,节省大量费用[18]。
ALSAQ-40(ALS自我评估问卷):指标有身体运动能力,饮食能力,社会交往能力,情绪反应。简单、可操作性强,可用于患者自我评价生活质量,便于信函随访[19]。
5.4、呼吸功能测定。建议用用力肺活量(FVC),FVC是最重要的生存率预测指标。Fallat发现FVC<50%预计值提示预后不良,Bensimon等在力如太临床试验中证实FVC可作为生存率预测指标,美国神经病学会及欧洲神经病学会联盟的ALS治疗指南均将其列为NIPPV的使用指征[12-13]。
5.5、生化指标。目前ALS尚无公认的用于诊断的特异性生物学标志物,也没有用于确定疾病进展的特异指标[20]。但有些研究已经提出肌酸激酶,肌酐,同型半胱氨酸,超敏C反应蛋白,抑胱素C,甚至尿酸,可能和疾病进展有关,值得临床进一步确定。
小结。ALS目前虽然没有治愈办法,但大量研究表明力如太,PEG,NIPPV及对症支持对延长患者存活期,提高生活质量有积极作用。明确诊断后,进一步细分临床表型,确定临床分期,客观的评估患者综合情况,有利于ALS患者合理选择治疗措施,也为新药的临床试验提供客观的评估指标。
转载:http://www.365heart.com/show/108267.shtml